New simulations help scientists understand how —and why — viruses spread
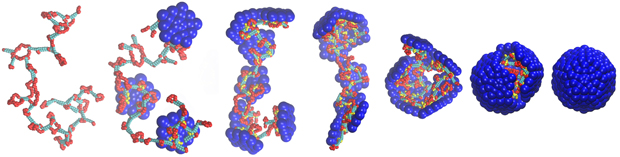
Above, snapshots from a computer simulation in which a model capsid assembles around a base-paired nucleic acid. The single-stranded nucleic acid is shown in red, the double-stranded nucleic acid in cyan, the capsid subunits are blue and the positive charges on the capsid proteins are shown in yellow.
This article is published By Ellen de Graffenreid
It’s not a hacker lab. At Brandeis University, sophisticated computational models and advances in graphical processing units are helping scientists understand the complex interplay between genomic data, virus structure and the formation of the virus’ outer “shell” — critical for replication.
“We hope that some of what we are finding will help researchers alter virus assembly, leaving viruses unable to replicate,” says post-doctoral fellow Jason Perlmutter, first author of thescientific paper describing the technique, published in the open access journal eLife.
Scientists know that many viruses are able to hijack the genetic machinery of host cells to produce copies of themselves and spread infection from cell to cell. For many virus families, a key part of this process is the formation of a protein “shell,” called a capsid, around the viral genome during the assembly process. [Representative movies]
The physics of this assembly process, which involves interactions between the negatively charged nucleic acid genome and the positively charged capsid protein, depends on a number of factors related to the structure of the virus genome.
“Changing all these critical genomic parameters in a live virus and looking at how capsid formation behaves is impossible given the speed of the process and our current imaging techniques,” says associate professor of physicsMichael Hagan, whose lab conducted the study.
That’s why the team’s modeling approach — the most realistic developed to date — is so important to scientists who are interested in how the virus capsid protein assembles around its genome in the cell. The Brandeis team used their modeling tool to calculate the optimal genome for a number of specific capsids — which vary in size, shape and surface properties.
“If you take the model and apply it to biological viruses, we are able to predict within a narrow range key structural features of the virus genome and, by extension, how these parameters control whether the capsid assembles or misassembles and what misassembly looks like,” says Perlmutter.
“Our tool should help scientists better understand the relationship between viral structure and packaging, making it easier to develop antiviral agents as well as aid the redesign of viruses for use in gene therapy and drug delivery.”
The work was supported by NIH NIAID grant number R01AI080791 and NSF XSEDE grant number TG-MCB090163.
Post a Comment
Thanks for reading my blog.
Note: only a member of this blog may post a comment.