Bioinformatics open source interoperability Hackathon at the Broad Institute
leave a comment »
Interoperability Hackathon
On April 7th and 8th, a group of biologists and programmers gathered at the Broad Institute to work on improving interoperability of open-source bioinformatics tools. Organized by the Open Bioinformatics Foundation and GenomeSpace team, this was part of the lead up to the Bioinformatics Open Source Conference (BOSC)
in July in Berlin. The event is part of an ongoing series of coding
sessions (Codefests or Hackathons) organized by the open bioinformatics
community, which give programmers who typically work together remotely a
chance to code and discuss in the same place for two days. These have
been successful in both producing new code and in building connections
which help sustain development of these community projects.
Goals and outcomes
One major challenge in analyzing biological data is interfacing
multiple bioinformatics tools. Tools often work independently, and where
general architectures like plugins or API exist they are often project
specific. This results in isolated islands of data exchange, but
transferring data or resources between tools requires work that is often
rate-limiting or insurmountable.
Our goal at the hackathon was to provide simple APIs and implementations that help facilitate transfers between multiple islands of functionality. GenomeSpace does this by providing a central hub and API to push and pull from tools. We wanted to generalize this to support multiple tools, and build client implementations that demonstrate this in practice. The long term goal is to encourage tool developers to provide server side APIs compatible with the more general library, making extension of the connector toolkit easier. For developers, the client API would allow them to easily transfer files between multiple tools without needing to learn and implement the specific transfer APIs of each tool.
We called this high level client library Genome Connector (gcon, for short) and took a practical approach by implementing client libraries that provide a common interface to multiple tools: GenomeSpace, Galaxy, BaseSpace, 23andMe and general key-value stores through jClouds. To identify a reasonable amount of work for two days, we focused on file transfer: authentication, finding files, getting and putting files to remote analysis platforms. In addition we defined some critical components for doing biological work:
The output of our discussion and coding are common Genome Connector implementations in multiple languages. GitHub repositories are available for in-progress Java, Python and Clojure implementations. These wrap multiple diverse tools and expose them through a common top level API, allowing developers to push and pull data from multiple tools.
I’m immensely grateful to the incredible participants who generously donated their time and expertise to help with these projects. For anyone interested we also have detailed documentation on discussions during the hackathon.
Our goal at the hackathon was to provide simple APIs and implementations that help facilitate transfers between multiple islands of functionality. GenomeSpace does this by providing a central hub and API to push and pull from tools. We wanted to generalize this to support multiple tools, and build client implementations that demonstrate this in practice. The long term goal is to encourage tool developers to provide server side APIs compatible with the more general library, making extension of the connector toolkit easier. For developers, the client API would allow them to easily transfer files between multiple tools without needing to learn and implement the specific transfer APIs of each tool.
We called this high level client library Genome Connector (gcon, for short) and took a practical approach by implementing client libraries that provide a common interface to multiple tools: GenomeSpace, Galaxy, BaseSpace, 23andMe and general key-value stores through jClouds. To identify a reasonable amount of work for two days, we focused on file transfer: authentication, finding files, getting and putting files to remote analysis platforms. In addition we defined some critical components for doing biological work:
- File metadata: We need to be able to store arbitrary key/value on objects to assign essential biological information necessary to interpret it, like organisms and genome build. In addition, metadata allows provenance and tracking of files by enabling annotation of files with history and processing steps.
- Filesets: Large biological files have secondary files with indexes, allowing indexed retrieval of data (for example: read bam and bai, variant vcf and idx, tabix gz and tbi). To avoid expensive reindexing, we want to group and transfer these together.
The output of our discussion and coding are common Genome Connector implementations in multiple languages. GitHub repositories are available for in-progress Java, Python and Clojure implementations. These wrap multiple diverse tools and expose them through a common top level API, allowing developers to push and pull data from multiple tools.
I’m immensely grateful to the incredible participants who generously donated their time and expertise to help with these projects. For anyone interested we also have detailed documentation on discussions during the hackathon.
Bioinformatics Open Source Conference
If you’re a bioinformatics programmers interested in open source
coding and helping answer biological questions by improving usability
and connectivity of tools, you’re welcome to join the OpenBio and BOSC
communities. The next BOSC conference is July 19th and 20th in Berlin, Germany as part of the ISMB conference. There will also be another two day Codefest proceeding BOSC on July 17th and 18th. Abstracts for talks at BOSC are due this Friday, April 12th. Looking forward to seeing everyone at future BOSC and coding events.
The influence of reduced resolution quality scores on alignment and variant calling
BAM file size reduction and quality score binning
We have a large upcoming whole genome sequencing project with Illumina, and they approached us about delivering BAM files with reduced resolution base quality scores. They have a white paper
describing the approach, which involves binning scores to reduce
resolution. This reduces the number of scores describing the quality of
a base from 40 down to 8.
The advantage of this approach is a significant reduction in file size. BAM files use BGZF compression, and the underlying gzip DEFLATE algorithm compresses based on shared text regions. Reducing the number of quality values increases shared blocks and improves compression. This reduces BAM file sizes by 25-35%: an exome BAM file reduced from 5.7Gb to 3.7Gb after quality binning.
The potential downside is that the reduction in quality resolution may impact alignment and variant calling approaches that rely on base quality scores. To assess this, I implemented quality score binning as part of the bcbio-nextgen analysis pipeline using the CRAM toolkit and ran alignment, recalibration, realignment and variant calling on:
The advantage of this approach is a significant reduction in file size. BAM files use BGZF compression, and the underlying gzip DEFLATE algorithm compresses based on shared text regions. Reducing the number of quality values increases shared blocks and improves compression. This reduces BAM file sizes by 25-35%: an exome BAM file reduced from 5.7Gb to 3.7Gb after quality binning.
The potential downside is that the reduction in quality resolution may impact alignment and variant calling approaches that rely on base quality scores. To assess this, I implemented quality score binning as part of the bcbio-nextgen analysis pipeline using the CRAM toolkit and ran alignment, recalibration, realignment and variant calling on:
- The original unbinned 40-resolution base quality BAM from an NA12878 exome.
- The BAM binned into 8-resolution base qualities before alignment.
- The BAM binned into 8-resolution base qualities before alignment and binned again following base quality score recalibration.
Alignment differences
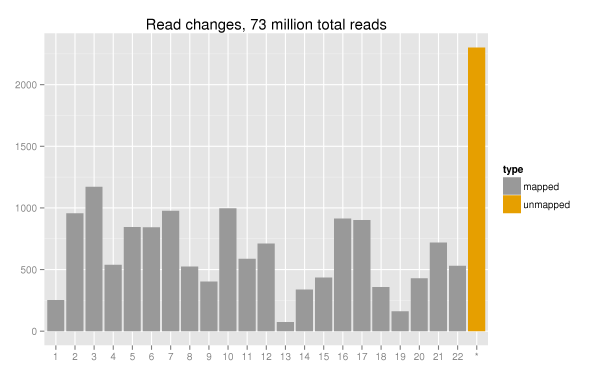
We aligned 100bp paired end reads with Novoalign,
a quality aware aligner. Comparison of mapped reads showed nearly no
impact on total mapped reads. The plot below shows a generic delta of
changes in mapped reads across the 22 autosomes alongside the increase
in unmapped pairs. Out of 73 million total reads, the changes account
for ~0.003% of the total reads. There also did not appear to be any
worrisome patterns of loss for specific chromosomes. Overall, there is a
minimal impact of quality score binning on the ability to align the
reads.
Variant call differences
We called variants using the GATK Unified Genotyper following the best practice recommendations for exomes
and then compared calls from original and binned quality scores. Both
approaches for binning — pre-binning, and pre-binning plus post-quality
recalibration binning — showed similar levels of concordance to
non-binned quality scores: 99.81 and 99.78, respectively. Since the
additional binning after recalibration provides a smaller prepared BAM
file for storage and has a similar impact to pre-binning only, we used
this for additional analysis of discordant variants.
The table below shows the discordant differences between the 40 quality score resolution and binned, 8 quality score resolution BAMs. 40 quality discordant variants are those called with full quality score resolution but not called, or called differently, after binning to 8 quality score resolution. Conversely, the 8-quality discordants are those called uniquely after quality binning:
We investigated the discordant variants further since 1.5% of the
total variant calls change as a result of binning, Of the 1851 unique
discordant variants, approximately half (928) fall into reproducible
variants identified by looking at ensemble combinations of replicates. Of these potentially problematic discordant variants more than half are in low coverage regions with less than 10 reads:
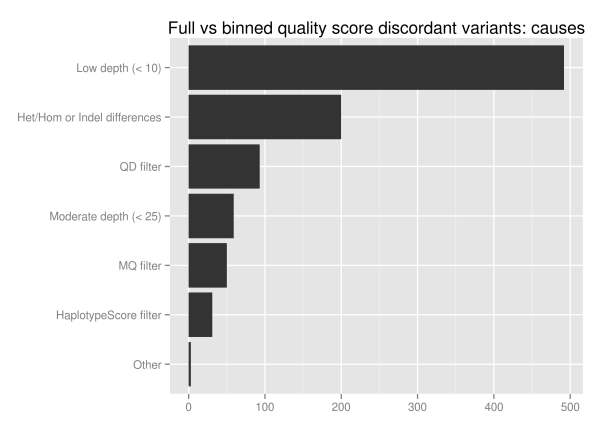
The major influence of quality score binning is resolution of variants in low coverage regions. This manifests as differences in heterozygote and homozygote calling, indel representation and filtering differences related to quality and mappability. To assess the potential impact, we looked at the loss in callable bases on a 30x whole genome sequence when moving from a minimum of 5 reads to a minimum of 10, using GATK’s CallableLoci tool. Regions with read coverage of 5 to 9 make up 4.7 million genome positions, 0.17% of the total callable bases.
In conclusion, quality score binning provides a useful reduction in
input file sizes with minimal impact on alignment. For variant calling,
use additional caution in low coverage regions with less than 10
supporting reads. Given the rapid increases in read throughput that are
driving the need for file size reduction, quality score binning is a
worthwhile tradeoff for high-coverage recalling work.
The table below shows the discordant differences between the 40 quality score resolution and binned, 8 quality score resolution BAMs. 40 quality discordant variants are those called with full quality score resolution but not called, or called differently, after binning to 8 quality score resolution. Conversely, the 8-quality discordants are those called uniquely after quality binning:
| Overall genotype concordance | 99.78 |
| concordant: total | 117887 |
| concordant: SNPs | 109144 |
| concordant: indels | 8743 |
| 40-quality discordant: total | 821 |
| 40-quality discordant: SNPs | 759 |
| 40-quality discordant: indels | 62 |
| 8-quality discordant: total | 1289 |
| 8-quality discordant: SNPs | 1240 |
| 8-quality discordant: indels | 49 |
| het/hom discordant | 259 |
The major influence of quality score binning is resolution of variants in low coverage regions. This manifests as differences in heterozygote and homozygote calling, indel representation and filtering differences related to quality and mappability. To assess the potential impact, we looked at the loss in callable bases on a 30x whole genome sequence when moving from a minimum of 5 reads to a minimum of 10, using GATK’s CallableLoci tool. Regions with read coverage of 5 to 9 make up 4.7 million genome positions, 0.17% of the total callable bases.
| 5 read minimum | 10 read minimum | |
|---|---|---|
| Callable bases | 2,775,871,235 | 2,771,109,000 |
| Percent callable | 96.90% | 96.73% |
| Low coverage | 17,641,980 | 22,404,215 |
| No coverage/ poor mapping | 71,272,008 | 71,272,008 |
An automated ensemble method for combining and evaluating genomic variants from multiple callers
Overview
A key goal of the Archon Genomics X Prize infrastructure is development of a set of highly accurate reference genome variants. I’ve described our work preparing these reference genomes,
and specifically defined the challenges behind merging genomic variant
calls from multiple technologies and calling methods. Comparing calls
from two different calling methods, for example GATK and samtools mpileup,
produces a large number of differing variants which need
reconciliation. Taking the overlapping subset from multiple callers is
too conservative and will miss real variations, while including all
calls is too liberal and introduces false positives.
Here I’ll describe a fully automated approach for preparing an accurate set of combined variant calls. Ensemble machine learning methods are a powerful way to incorporate inputs from multiple models. We use a heuristic and support vector machine (SVM) algorithm to consolidate variants, producing a final set of calls with better sensitivity and specificity than current best practice methods. The approach is open source, fully automated and generalizable to both human diploid sequencing as well as X Prize haploid reference fosmids.
We use a pair of replicates from EdgeBio’s clinical exome sequencing pipeline to prepare ensemble variant calls in the widely studied HapMap NA12878 genome. Compared to variants from a single calling method, the ensemble method produced more concordant variants when comparing the replicates, with fewer discordants. The finalized ensemble calls also provide a useful method to compare strengths and weaknesses of individual calling methods. The implementation is freely available and I’ll discuss how to get it running on your data so you can use, critique and extend the methods. This work is a collaboration between Harvard School of Public Health, EdgeBio and NIST.
Here I’ll describe a fully automated approach for preparing an accurate set of combined variant calls. Ensemble machine learning methods are a powerful way to incorporate inputs from multiple models. We use a heuristic and support vector machine (SVM) algorithm to consolidate variants, producing a final set of calls with better sensitivity and specificity than current best practice methods. The approach is open source, fully automated and generalizable to both human diploid sequencing as well as X Prize haploid reference fosmids.
We use a pair of replicates from EdgeBio’s clinical exome sequencing pipeline to prepare ensemble variant calls in the widely studied HapMap NA12878 genome. Compared to variants from a single calling method, the ensemble method produced more concordant variants when comparing the replicates, with fewer discordants. The finalized ensemble calls also provide a useful method to compare strengths and weaknesses of individual calling methods. The implementation is freely available and I’ll discuss how to get it running on your data so you can use, critique and extend the methods. This work is a collaboration between Harvard School of Public Health, EdgeBio and NIST.
Comparison materials and algorithm
A difficult aspect of evaluating variant calling methods is
establishing a reference set of calls. For X Prize we use three
established methods, each of which comes with tradeoffs. Metrics like transition/transversion ratios or dbSNP overlap
provide a global picture of calling but are not fine grained enough to
distinguish improvements over best practices. Sanger validation
restricts you to a manageable subset of calls. Comparisons against
public resources like 1000 genomes bias results towards technologies and callers used in preparing those variant callsets.
Here we employ a fourth method by comparing replicates from EdgeBio’s clinical exome sequencing pipeline. These are NA12878 samples independently prepared using Nimblegen’s version 3.0 kit and sequenced on an Illumina HiSeq. By comparing the replicates in regions with 4 or more reads in both samples, we identify the ability of variant calling algorithms to call identical variations with differing coverage and error profiles.
We aligned reads with novoalign and performed deduplication, base recalibration and realignment using GATK best practices. With these prepared reads, we called variants with five approaches:
The first filtering step is to heuristically identify trusted variants based on the number of callers supporting them. This configurable parameter allow you to make an initial conservative cutoff for including variants in the final calls: I trust variants supported by N or more callers.
For the remaining calls that fall below the trusted support cutoff, we distinguish true and false positives using a support vector machine (SVM). The annotated metrics described above are the input parameters and we prepare true and false positives for the classifier using a multi-step process:
Here we employ a fourth method by comparing replicates from EdgeBio’s clinical exome sequencing pipeline. These are NA12878 samples independently prepared using Nimblegen’s version 3.0 kit and sequenced on an Illumina HiSeq. By comparing the replicates in regions with 4 or more reads in both samples, we identify the ability of variant calling algorithms to call identical variations with differing coverage and error profiles.
We aligned reads with novoalign and performed deduplication, base recalibration and realignment using GATK best practices. With these prepared reads, we called variants with five approaches:
- GATK UnifiedGenotyper – Bayesian approach to call SNPs and indels, treating each position independently.
- GATK HaplotypeCaller – Performs local de-novo assembly to call SNPs and indels on individual haplotypes.
- FreeBayes – Bayesian calling approach that handles simultaneous SNPs and indel calling via assessment of regional haplotypes.
- samtools mpileup – Uses an approach similar to GATK’s UnifiedGenotyper for SNP and indel calling.
- VarScan – Calls variants using a heuristic/statistic approach eliminating common sources of bias.
The first filtering step is to heuristically identify trusted variants based on the number of callers supporting them. This configurable parameter allow you to make an initial conservative cutoff for including variants in the final calls: I trust variants supported by N or more callers.
For the remaining calls that fall below the trusted support cutoff, we distinguish true and false positives using a support vector machine (SVM). The annotated metrics described above are the input parameters and we prepare true and false positives for the classifier using a multi-step process:
- Use variants found in all callers as the true positive set, and variants found in a single caller as false positives. Use these training variants to identify an initial set of below-cutoff variants to include and exclude.
- With this initial set of below-cutoff true/false variants, re-train multiple classifiers stratified based on variant characteristics: variant type (indels vs SNPs), zygosity (heterozygous vs homozygous) and regional sequence complexity.
- Use these final classifiers to identify included and excluded variants falling below the trusted calling support cutoff.
Ensemble calling improvements
We assess calling sensitivity and specificity by comparing
concordant and discordant variant calls between the replicates. To
provide a consistent method to measure both SNP and indel correctness,
we use the positive predictive value
as the percentage of concordant calls between duplicates (concordant
variants / (concordant variants + discordant variants)). This is
different than the overall concordance rate, which also includes
non-variant regions where both replicates do not call a variation. As a
result these percentages will be lower if you expect the 99% values
that result when including reference calls. The advantage of this metric
is that it’s easily interpreted as the percentage of concordant called
variants. It also allows individual comparisons of SNPs and indels,
since the overall number of indels are low compared to the total bases
considered. GATK’s VariantEval documentation has a nice discussion of alternative metrics to genotype concordance.
As a baseline we used calls from GATK’s UnifiedGenotyper to represent a current best practice approach. GATK calls 117079 SNPs, 86.6% of which are concordant. It also calls 14966 indels, with 64.6% concordant. Here are the full concordant and discordant numbers, broken down by variant type and replicate:
Our ensemble method produces improvements in both total concordant
variants detected and the ratio of concordant to discordants. For SNPs,
the ensemble calls add 5345 additional variants to a total of 122424,
with an increase in concordance to 87.4%. For indels the major
improvement is in removal of discordants: We identify 14184 indels with
67.2% concordant. Here is the equivalent table for the ensemble method:
For scientists who have worked to increase sensitivity and
specificity of individual variant callers, it’s exciting to be able to
improve both simultaneously. As mentioned above, you can also tune the
method to increase specificity or sensitivity by adjusting the support
needed for including trusted variants.
The final ensemble callsets from both replicates are available as VCF files from GenomeSpace in the
As a baseline we used calls from GATK’s UnifiedGenotyper to represent a current best practice approach. GATK calls 117079 SNPs, 86.6% of which are concordant. It also calls 14966 indels, with 64.6% concordant. Here are the full concordant and discordant numbers, broken down by variant type and replicate:
| concordant: total | 111159 |
| concordant: SNPs | 101495 |
| concordant: indels | 9664 |
| rep1 discordant: total | 9857 |
| rep1 discordant: SNPs | 7514 |
| rep1 discordant: indels | 2343 |
| rep2 discordant: total | 11029 |
| rep2 discordant: SNPs | 8070 |
| rep2 discordant: indels | 2959 |
| het/hom discordant | 4181 |
| concordant: total | 116608 |
| concordant: SNPs | 107063 |
| concordant: indels | 9545 |
| rep1 discordant: total | 9555 |
| rep1 discordant: SNPs | 7581 |
| rep1 discordant: indels | 1974 |
| rep2 discordant: total | 10445 |
| rep2 discordant: SNPs | 7780 |
| rep2 discordant: indels | 2665 |
| het/hom discordant | 3975 |
The final ensemble callsets from both replicates are available as VCF files from GenomeSpace in the
xprize/NA12878-exome-v_03 folder: - NA12878 exome ensemble calls, replicate 1 – Variants (VCF), Callable regions (BED)
- NA12878 exome ensemble calls, replicate 2 – Variants (VCF), Callable regions (BED)
Comparison of calling methods
Calling the same samples with multiple callers allows direct
comparisons between calling methods. The advantage of producing an
accurate final set of ensemble calls is that this provides a baseline to
evaluate the strengths and weaknesses of different calling methods. The
figure below compares concordant, missing variants and additional
variants called by each of the 5 methods in comparison with the
consolidated ensemble calls:
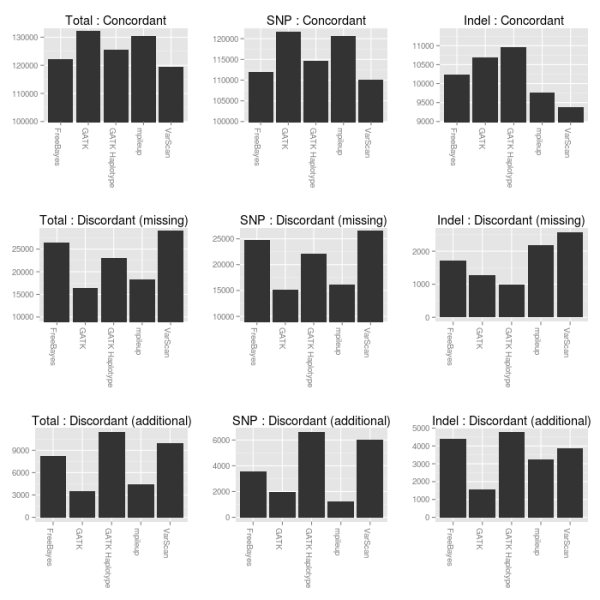
- GATK UnifiedGenotyper provides the best SNP calling, followed closely by samtools mpileup.
- For indel calling, the GATK HaplotypeCaller produces the most concordant calls followed by UnifiedGenotyper and FreeBayes. UnifiedGenotyper does good as well, but is conservative and has the fewest additional indels. FreeBayes and GATK HaplotypeCaller both provide resolution of individual haplotypes which helps in regions with heterozygous indels or closely spaced SNPs and indels.
- If you want to use a single variant caller, GATK UnifiedGenotyper does the best overall job.
- If you wanted to choose free open-source tools for calling, I would recommend samtools for SNP calling and FreeBayes for indel calling.
Availability and usage
Combining multiple calling approaches improves both sensitivity and
specificity of the final set of variants. The downside is the need to
run and coordinate calls from all of the different callers. To mitigate
this, we developed an automated pipeline that ties together multiple
open-source tools using two custom components:
EdgeBio kindly made the NA12878 datasets used in this analysis publicly available:
I welcome feedback on the approach, data or tools and am actively working to extend this and make it easier to use. As re-sequencing becomes increasingly important for human health applications it is critical that we develop open, shared best-practice workflows to handle the data processing. This allows us to focus back on the fun and difficult work of understanding the biology.
- bcbio-nextgen – A Python framework to run a full sequencing analysis pipeline from input fastq files to consolidated ensemble variant calls. It supports multiple aligners and variant callers, and enables distributed work over multiple cores on a large machine or multiple machines in a cluster environment.
- bcbio.variation – A Clojure toolkit build on top of GATK’s variant API that provides ensemble call preparation as well as more general functionality for normalizing and comparing variants produced by multiple callers.
wget https://raw.github.com/chapmanb/bcbio-nextgen/master/scripts/bcbio_nextgen_install.py
python bcbio_nextgen_install.py install_directory data_directory
With the dependencies installed, you describe the input files and analysis with a YAML formatted input file. The NA12878 ensemble configuration file used for this analysis provides a useful starting point. Run the analysis, distributed on multiple cores, with: bcbio_nextgen.py bcbio_system.yaml ensemble_sample.yaml -n 8
The bcbio-nextgen documentation
provides additional details about configuration inputs and distributed
processing. The framework generally handles the automation and
processing involved with high throughput sequencing analysis. EdgeBio kindly made the NA12878 datasets used in this analysis publicly available:
I welcome feedback on the approach, data or tools and am actively working to extend this and make it easier to use. As re-sequencing becomes increasingly important for human health applications it is critical that we develop open, shared best-practice workflows to handle the data processing. This allows us to focus back on the fun and difficult work of understanding the biology.
Genomics X Prize public phase update: variant classification and de novo calling
Background
Last month I described our work at HSPH and EdgeBio preparing reference genomes for the Archon Genomics X Prize
public phase, detailing methods used in the first version of our
NA19239 variant calls. We’ve been steadily improving the calling
approaches, and released version 0.2 on the X Prize validation website and GenomeSpace. Here I’ll describe the improvements we’ve made over the last month, focusing on two specific areas:
As a reminder, all of the code and data used here is freely available:
- De novo calling: Zam Iqbal suggested using his cortex_var de novo variant caller in addition to the current GATK, FreeBayes and samtools callers. With his help, we’ve included these calls in this release, and provide comparisons between de novo and alignment based methods.
- Improved variant classification: Consolidating variant calls from multiple callers involves making tough choices about when to include or exclude variants. I’ll describe the details of selecting metrics for use in SVM classification and filtering of variants.
As a reminder, all of the code and data used here is freely available:
- The variant analysis infrastructure, built on top of GATK, automates genome preparation, normalization and comparison. It provides a full pipeline, driven by simple configuration files, for consolidating multiple variant calls.
- The combined variant calls, including training data and potential true and false positives, are available from GenomeSpace:
Public/chapmanb/xprize/NA19239-v0_2. - The individual variant calls for each technology and calling method are also available from GenomeSpace:
Public/EdgeBio/PublicData/Release1.
de novo variant calling with cortex_var
de novo variant calling performs reference-free assembly of
either local or global genome regions, then subsequently uses these
assemblies to call variants relative to a known reference. The advantage
is that assemblies can avoid errors associated with mapping to the
reference, helping resolve large variations as well as small variations
near problem alignments or low complexity regions.
Hybrid approaches that use localized de novo assembly in variant regions help mitigate the extensive computational requirements associated with whole-genome assembly. Complete Genomics variant calling and GATK 2.0′s Haplotype Caller both provide pipelines for hybrid de novo assembly in variant detection. The fermi and SGA assemblers are also used in variant calling, although the paths from assembly to variants are not as automated.
Thanks to Zam’s generous assistance, we used cortex_var for localized de novo assembly and variant calling within individual fosmid boundaries. As a result, CloudBioLinux now contains automated build instructions for cortex_var , handling binary builds for multiple k-mer and color combinations. An automated cortex_var pipeline, part of the bcbio-nextgen toolkit, runs the processing workflow:
11% of the GATK calls and 14% of the cortex_var calls are
discordant. The one area where cortex_var does especially well is on
indels: 19% of the cortex_var indels do not agree with GATK, in
comparison with 37% of the GATK calls and 25% of the samtools calls. The
current downside to this is SNP calling, where cortex_var has 3 times
more discordant calls than GATK.
Hybrid approaches that use localized de novo assembly in variant regions help mitigate the extensive computational requirements associated with whole-genome assembly. Complete Genomics variant calling and GATK 2.0′s Haplotype Caller both provide pipelines for hybrid de novo assembly in variant detection. The fermi and SGA assemblers are also used in variant calling, although the paths from assembly to variants are not as automated.
Thanks to Zam’s generous assistance, we used cortex_var for localized de novo assembly and variant calling within individual fosmid boundaries. As a result, CloudBioLinux now contains automated build instructions for cortex_var , handling binary builds for multiple k-mer and color combinations. An automated cortex_var pipeline, part of the bcbio-nextgen toolkit, runs the processing workflow:
- Start with reads aligned to fosmid regions using novoalign.
- For each fosmid region, extract all mapped reads along with a local reference genome for variant calling.
- de novo assemble all reads in the fosmid region and call variants against the local reference genome using cortex_var’s Bubble Caller.
- Remap regional variant coordinates back to the full genome.
- Combine all regional calls into the final set of cortex_var calls.
| concordant: total | 153787 |
| concordant: SNPs | 130913 |
| concordant: indels | 22874 |
| GATK discordant: total | 20495 |
| GATK discordant: SNPs | 6522 |
| GATK discordant: indels | 13973 |
| cortex_var discordant: total | 26790 |
| cortex_var discordant: SNPs | 21342 |
| cortex_var discordant: indels | 5448 |
